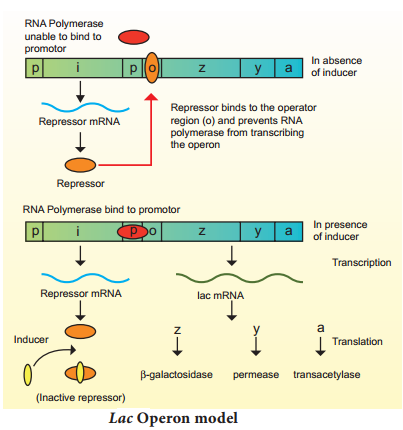
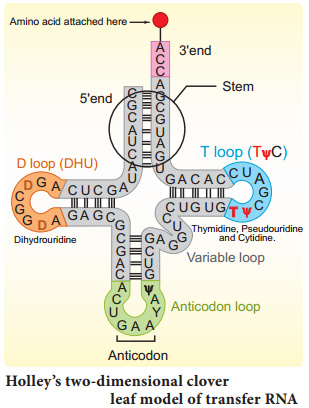
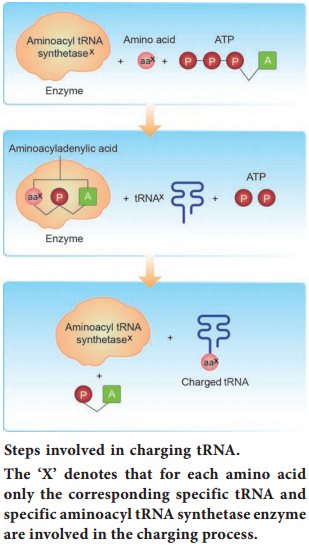
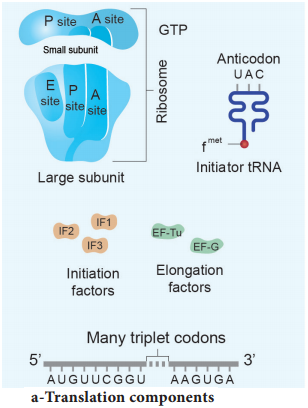
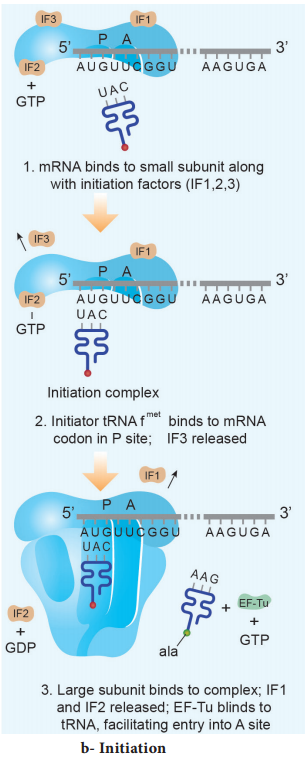
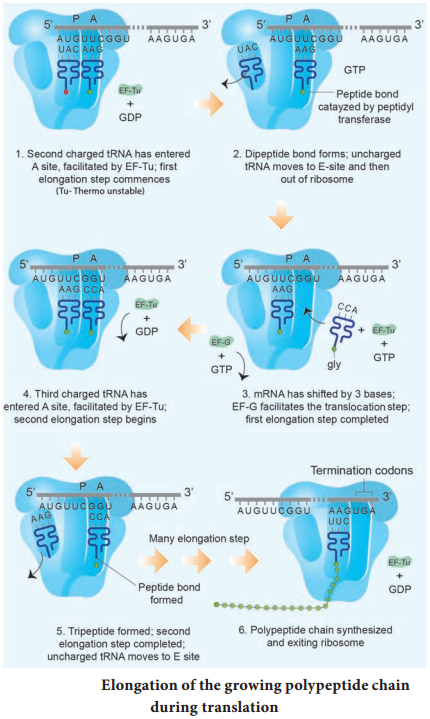
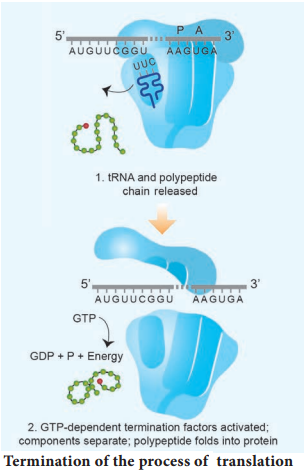
Learninsta presents the core concepts of Biology with high-quality research papers and topical review articles.
DNA Finger Printing Technique
The DNA fingerprinting technique was first developed by Alec Jeffreys in 1985 (Recipient of the Royal Society’s Copley Medal in 2014). Each of us have the same chemical structure of DNA. But there are millions of differences in the DNA sequence of base pairs. This makes the uniqueness among us so that each of us except identical twins is different from each other genetically.
The DNA of a person and finger prints are unique. There are 23 pairs of human chromosomes with 1.5 million pairs of genes. It is a well known fact that genes are segments of DNA which differ in the sequence of their nucleotides.
Not all segments of DNA code for proteins, some DNA segments have a regulatory function, while others are intervening sequences (introns) and still others are repeated DNA sequences. In DNA fingerprinting, short repetitive nucleotide sequences are specific for a person. These nucleotide sequences are called as variable number tandem repeats (VNTR).The VNTRs of two persons generally show variations and are useful as genetic markers.
DNA figer printing involves identifying diffrences in some specific regions in DNA sequence called repetitive DNA, because in these sequences, a small stretch of DNA is repeated many times. These repetitive DNA are separated from bulk genomic DNA as different peaks during density gradient centrifugation. The bulk DNA forms a major peak and the other small peaks are referred to as satellite DNA.
Depending on base composition (A : T rich or G : C rich), length of segment and number of repetitive units, the satellite DNA is classified into many sub categories such as micro-satellites, minisatellites, etc., These sequences do not code for any proteins, but they form a large portion of human genome.
These sequences show high degree of polymorphism and form the basis of DNA figerprinting (Fig. 5.15). DNA isolated from blood, hair, skin cells, or other genetic evidences lef at the scene of a crime can be compared through VNTR patterns, with the DNA of a criminal suspect to determine guilt or innocence. VNTR patterns are also useful in establishing the identity of a homicide victim, either from DNA found as evidence or from the body itself.
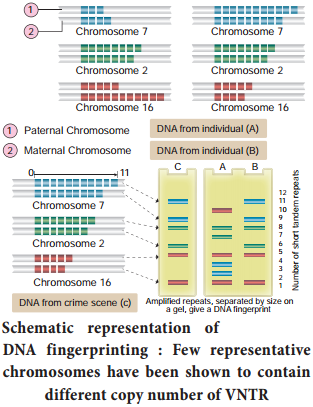
The Steps in DNA Fingerprinting technique is depicted in Fig. 5.16.
1. Extraction of DNA
The process of DNA fingerprinting starts with obtaining a sample of DNA from blood, semen, vaginal fluids, hair roots, teeth, bones, etc.,
2. Polymerase chain reaction (PCR)
In many situations, there is only a small amount of DNA available for DNA figerprinting. If needed many copies of the DNA can be produced by PCR (DNA amplifiation).
3. Fragmenting DNA
DNA is treated with restriction enzymes which cut the DNA into smaller fragments at specific sites.
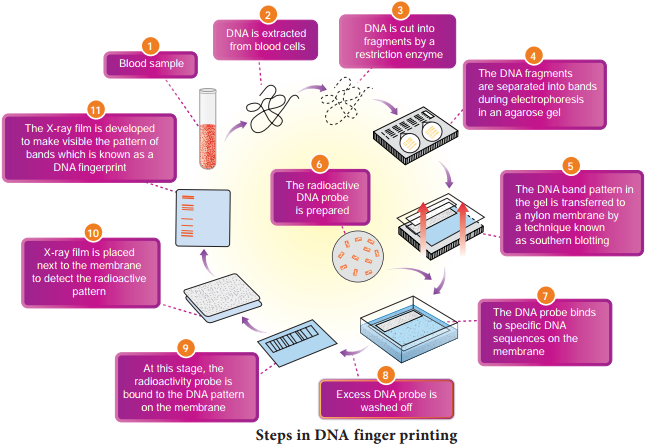
4. Separation of DNA by electrophoresis
During electrophoresis in an agarose gel, the DNA fragments are separated into bands of different sizes. The bands of separated DNA are sieved out of the gel using a nylon membrane (treated with chemicals that allow for it to break the hydrogen bonds of DNA so there are single strands).
5. Denaturing DNA
The DNA on gels is denatured by using alkaline chemicals or by heating.
6. Blotting
The DNA band pattern in the gel is transferred to a thin nylon membrane placed over the ‘size fractionated DNA strand’ by Southern blotting.
7. Using probes to identify specific DNA
A radioactive probe (DNA labeled with a radioactive substance) is added to the DNA bands. The probe attaches by base pairing to those restriction fragments that are complementary to its sequence. The probes can also be prepared by using either ‘florescent substance’ or ‘radioactive isotopes’.
8. Hybridization with probe
After the probe hybridizes and the excess probe washed off a photographic film is placed on the membrane containing ‘DNA hybrids’.
9. Exposure on fim to make a genetic/DNA Fingerprint
The radioactive label exposes the film to form an image (image of bands) corresponding to specific DNA bands. The thick and thin dark bands form a pattern of bars which constitutes a genetic fingerprint.
Application of DNA figer printing
Forensic analysis:
1. It can be used in the identification of a person involved in criminal activities, for settling paternity or maternity disputes, and in determining relationships for immigration purposes.
2. Pedigree analysis – inheritance pattern of genes through generations and for detecting inherited diseases.
3. Conservation of wild life – protection of endangered species. By maintaining DNA records for identification of tissues of the dead endangered organisms.
4. Anthropological studies – It is useful in determining the origin and migration of human populations and genetic diversities.